How To Perform Aseptic Media Fill Simulations
What are aseptic processes?
Aseptic processes are methods or procedures that are undertaken in a sterile environment. The aseptic sterile environment is maintained through specialized equipment that prevents microbial material from technicians, raw materials, or machinery from contaminating medical devices or products.
The terms aseptic and sterile are not synonymous. While both sterile and aseptic products will prevent microbial contamination following use, the processes by which microbial contamination is prevented are different. The term sterile means a complete absence of viable microorganisms or microbes that have the potential to reproduce. Thus, sterile products are often chemically or heat sterilized after being placed in their final packaging. The chemical or heat sterilization kills any microorganisms inside the products (obtained during manufacturing and packaging). This chemical or heat sterilization process after final product packaging is known as terminal sterilization. However, an aseptic process prevents contamination by the exclusion of microorganisms. Though the definitions for aseptic and sterile are not the same, sterile is used interchangeably with aseptic. Indeed, many products labeled as sterile are manufactured by aseptic processing rather than terminal sterilization. Aseptic processes can vary in complexity from comparatively simple filling-sealing to lengthy manufacturing sequences for complex items, such as medical devices.
Why are aseptic media fill simulations important for aseptic products?
Stability is a suspended state where there is no change. In terms of FDA regulations, stability covers five FDA categories: chemical stability, physical stability, therapeutic stability, toxicological stability, and microbiological stability of a medical device or product. Chemical stability refers primarily to drug formulations and ensures that all molecules in a formulation remain in their therapeutic state and do not undergo additional chemical reactions over time. However, medical devices must also be protected from oxidative chemical reactions or other chemical reactions that could cause the degradation of product materials. Physical stability refers to biological therapeutics (maintaining the physical integrity of protein structures over time) and the physical functional stability of medical devices or combination products. Therapeutic stability refers to the stability of a product or device’s therapeutic efficacy over time. Toxicological stability refers to the stability of the medical product, cosmetic, drug, or medical device’s toxicity levels over time. Finally, microbiological stability is the sterility of the product over time.
FDA Observations Related To Media Fill Processes
- Inadequate investigation of media fill failure.
- Inadequate training of employees after media fill failure.
- Media fills did not follow SOP.
- Media fill aborted due to high particulate counts, but an inadequate investigation into reasons for high counts.
- Media fill did not start at the point after the product had been sterilized.
- Defective vials were discarded prior to incubation and not counted as failures.
- The number of units filled is too small.
- Media fills did not simulate what was documented in batch records.
- Specific environmental data not collected during fill.
With a multitude of factors affecting the sterility of aseptic processes, the use of sterile culture media has become the best way to quantitatively validate that all the aseptic processing factors are functional to create a sterile product. Culture media replaces the prepared, filtered, and filled into the final container for media fill studies.
Culture media will pick up any microbes found in the production process and allow them to grow. Thus, any breach of asepsis in the manufacturing process, components, personnel involved, and equipment used will show up as positive microbial growth in the culture media. As mentioned earlier, the FDA is strict about media fill simulations being designed to simulate all aspects of the actual aseptic process accurately. Factors to consider for designing your media fill study to support meeting FDA and EU guidelines are listed below.
Factors to Consider In the Design of Media Fill Studies
- Duration of longest run
- Worst-case environmental conditions
- Number and type of interventions, stoppages, adjustments, transfers
- Aseptic assembly of equipment
- Number and activities of personnel
- Number of aseptic additions
- Shift breaks, changes, multiple gowning
- Number/type of aseptic equipment disconnections and connections
- Aseptic samples
- Line speed/configuration
- Manual weight checks
- Operator fatigue
- Container/closure types run on the line
- Temperature/relative humidity extremes
- Conditions permitted before line clearance
- Container/closure surfaces that contact formulation during the aseptic process

How do you perform an aseptic process simulation (media fill simulation) test?
The FDA advocates videotaping media fills to identify any improvements in personnel practices for the aseptic processes. The media fill simulations (also known as process simulation tests) use sterile trypticase soy broth under conditions simulating (as closely as possible) the manufacturing, processing, and filling requirements of a product. Media fill simulations use an entire lot of media, traditionally 4750 units. After the sterile media is processed through the planned aseptic conditions of the products, prior to incubation, all filled units are inverted or swirled to enable the media to contact all internal surfaces of the filled containers. Then the entire media lot is incubated at temperatures to support microbial growth, usually rotating 20–25◦C storage and 30–35◦C storage, for at least 14 days before being examined for the appearance of microbial growth. Positive controls and negative controls prove the microbial growth of the media. The sterility of the media must be examined for these studies. For positive controls, the challenge organisms used should be organisms isolated from the manufacturing environment and personnel monitoring. The positive control units are inoculated with approximately 100 colony-forming units (CFUs) of these challenge organisms. After the 14-day incubation period of the media fill containers, negative control units are inoculated with challenge organisms to prove that the media will still support microbial growth after the 14-day incubation period. Trained qualified inspectors evaluate the filled units before and after incubation. Any filled unit with a container integrity issue (a leak, tear, etc.) will be excluded from the media fill incubation, just as a filled product vial would be rejected if a critical defect were found. However, if a media-filled unit is only found damaged after incubation is underway, it must remain incubated and counted in the data for the media fill batch. To pass a media fill simulation test, not more than 0.1% of the challenged units in the media fill lot may show microbial growth. Ideally, the media growth requirements are “approaching zero.” Criteria for media fill simulation tests are further described below.
When should media fill simulations be performed?
The media fill simulations are a “one-time” evaluation of the aseptic processing operation. Media fill simulations are performed when a new filling line or new product container is introduced to an aseptic process. For the initial qualification of a fill line or product, three consecutive, separate, and successful media fill runs are required. The industry standard for each consecutive run is 4750 fills per run. A “successful” run is where no microbial growth is present in any of the units filled with sterile broth. All personnel involved in the aseptic filling (operators, maintenance personnel, microbiology support personnel) must participate in at least one media fill run per year to cover any personnel-to-personnel variability during aseptic processing. For each filling line and process, the filling operation will be validated from the smallest to the largest container size that will be used for the validation.
After initial qualification, media fills are then conducted usually twice a year on the same filling line to assure the maintenance of aseptic conditions over time. For periodic qualification, only one successful media fill run is required. If any media fill run fails or significant changes occur with the line, facility, or personnel, the initial qualification media fill (three consecutive successful runs) must be performed. Any changes in the aseptic process must be evaluated for microbial ingress risk to see if the modification necessitates a full media fill validation run. Any media fill failure must be thoroughly investigated and followed by multiple repeat media fill runs. Media fill runs cannot be “invalidated.” The number of containers filled with media is ideally the same as the actual number filled for the validated product. The number of units filled for media fill testing must be sufficient to reflect the effects of the “worst-case” situation for filling. Thus, a representative “worst-case” number for large batch sizes can be used instead of the full batch size amount.
What are the acceptance criteria for media fill simulation tests?
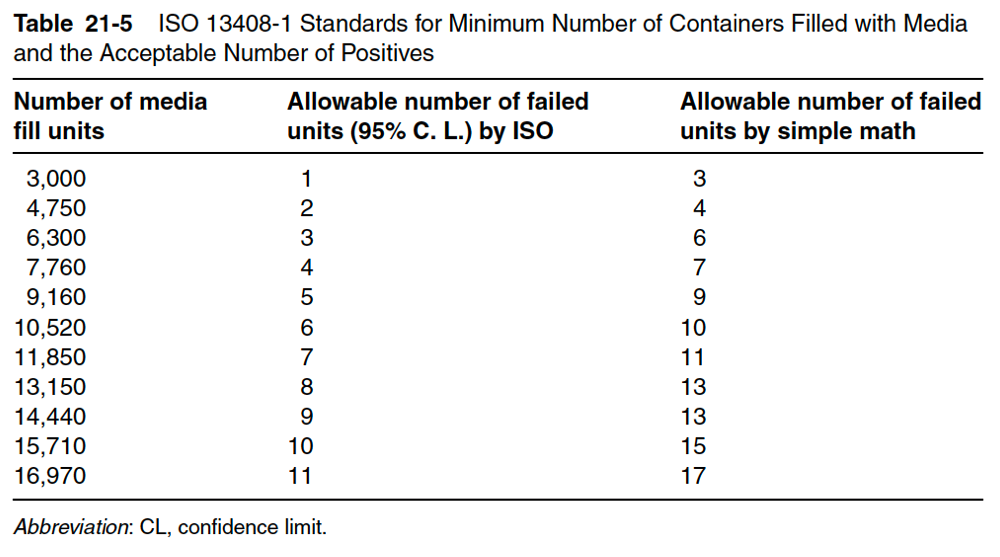
When media filling first started, the acceptable rate of positives (containers with microbial growth after aseptic processing) was 1 out of 1000 (0.1%). Later that number became 1 out of 3000 to account for 95% confidence of a contamination rate of 0.1%. Currently, a single positive out of 4750 is preferred. Table 21-5 details the International Standards Organization (ISO) standard, which is used to determine the minimum number of containers filled with media per lot and the minimum acceptable number of positives for that number of containers. Note that any positives in an aseptic process media fill test cause investigation of a microbial breach in the current manufacturing or filling process. As mentioned earlier, the industry standard for initial performance qualification of a new product, filling line, or closing line is three consecutive runs of 4750 units. Most media fills are a minimum of 3 hours; some may be as long as 24 hours. A fill of 4750 units is used for routine semiannual re-qualification tests. For these tests, the expected number of positive media fills (growth seen upon incubation) is zero. One or more failures mean a significant breach in the aseptic manufacturing process and require an investigation.
Summary
Overall, an aseptic process prevents contamination by the exclusion of microorganisms. Media fill simulations are one of three main components to aseptic process validation and equipment qualification for aseptic processes. During a media fill simulation, sterile growth media is processed through the entire aseptic processing sequence and then tested after the filling process for microbial growth. If microbial growth is found, the aseptic process must be further modified to exclude microorganisms. If the sterile growth media remains sterile after aseptic processing, the aseptic process passes one of three aseptic process validation steps. Consider MycoScience when selecting a contract manufacturing organization to support you with an aseptic syringe and vial filling.
MycoScience is a contract manufacturing organization that specializes in filling sterile syringes and vials for parenteral products. MycoScience also offers Sterilization Validations, Bacterial Endotoxin Testing, Preservative Efficacy Testing, Bioburden Testing, Cleaning Validations, Microbial Aerosol Challenge Testing, Accelerated Aging, Microbiology Testing, Cytotoxicity Testing, EO Residual Testing, Package Integrity Testing & Environmental Monitoring services for medical device companies, and allied industries. MycoScience is an ISO 13485 certified facility.
References
Michael J. Akers. Sterile Drug Products Formulation, Packaging, Manufacture, and Quality. Drugs and the Pharmaceutical Sciences. Informa Healthcare. 2010.
Sharing this in your social netwroks