Preservative Challenge Testing vs. Bacterial Endotoxin Testing
What is preservative challenge testing (preservative efficacy testing)?
Preservative challenge testing, also known as preservative efficacy testing (PET), determines the effectiveness of a preservative during its shelf life and evaluates how well a product withstands microbial contamination during use. Preservatives are substances mixed into creams, gels, and other liquids. Preservatives are used to prevent the growth of bacteria, fungi, and other microorganisms within a medical, cosmetic, household, or food product. Preservatives also help keep the freshness of the appearance of a product and keep its consistency intact over time.
What is a bacterial endotoxin?
Endotoxins come from the cell walls of gram-negative bacteria. The endotoxins themselves are molecules with fat components and complex sugar components (also known as polysaccharides). Therefore, endotoxins are also known as lipopolysaccharides (LPS) in scientific literature. Endotoxins are also considered pyrogens because they trigger the innate immune system and produce fever when released within the human body.
What is bacterial endotoxin testing?
A bacterial endotoxins test (BET) uses an assay known as the Limulus Amoebocyte Lysate (LAL) test. BET testing is considered a pyrogenicity test. However, do not confuse BET testing with rabbit pyrogen testing. LAL is an extract of blood cells from the Atlantic horseshoe crab. LAL detects the LPS of the cell wall of gram-negative bacteria, even if these bacteria are dead. LAL detects LPS through clotting and gelling in the presence of LPS, allowing for precise calculations to be made as to the concentration of endotoxins in a sample.
What products require preservative challenge testing?
Preservative challenge testing can be used to determine the best preservative to use in your topical formulation and the minimum effective concentration needed to preserve the pharmaceutical, food, or biotechnology product being assessed. Topical formulations (gels, creams, ointments, and lotions) comprise a water or oil base that delivers various medications, natural substances (such as herbs), or moisture through the skin. Topical cosmetics contain preservatives to support the stability of the formulation and prevent microbial growth during repeated product use. Preservative challenge testing is also used for parenteral products containing multiple doses. In these products, antimicrobial preservatives inhibit the growth of any microorganisms introduced during repeated insertion and withdrawal to load individual amounts.
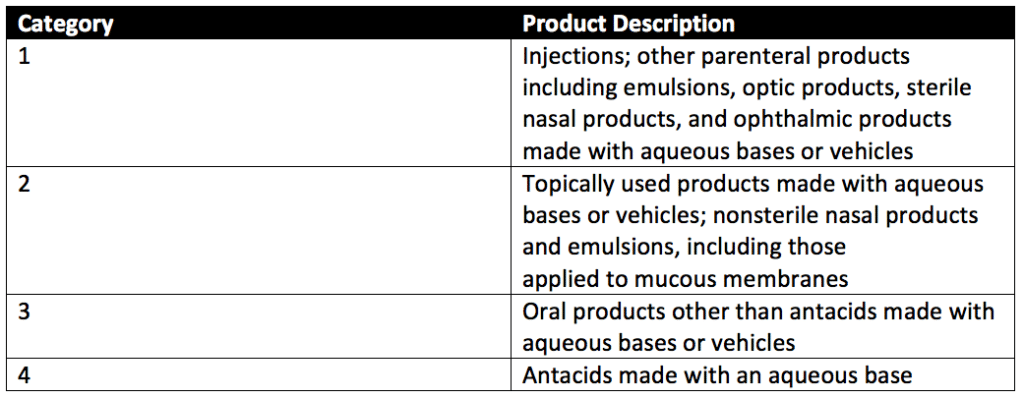
Preservative efficacy testing can evaluate four categories of products with preservative challenge testing. These product categories are detailed in Table 1 of USP 51, reproduced below.
What products require bacterial endotoxin testing?
Cosmetics do not require bacterial endotoxin testing, but FDA-regulated medical products (such as medical devices, parenteral products, and combination products) require BETs. Bacterial endotoxin testing is an important quality control step that detects product endotoxin contamination levels at any production stage, from initial product manufacture to final distribution. As microorganisms exist on every surface (including our body), bacterial endotoxins can be accidentally introduced during the manufacturing or packaging process in many ways. Some of the most common examples are contamination through the raw materials used, technicians, tubing/piping used to transfer product between development stages in a process, or the manufacturing environment itself. With such abundant sources of contamination, regular endotoxin testing supports the long-term control of manufacturing sites.
How is preservative challenge testing performed?
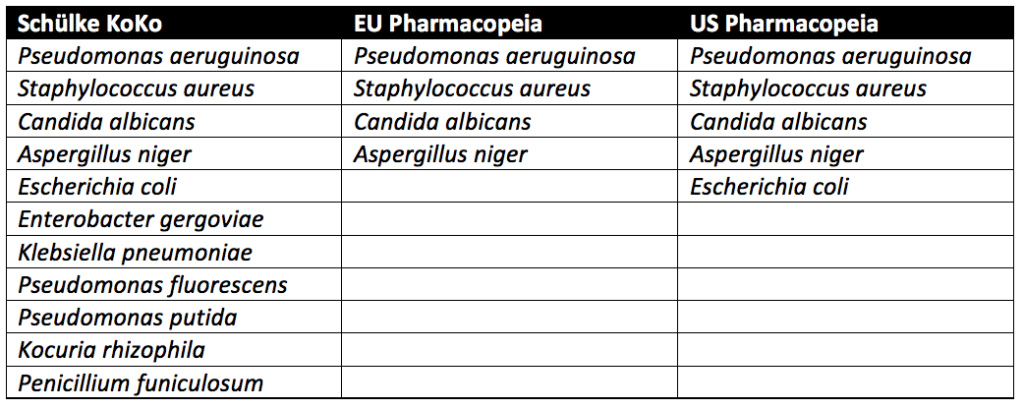
Three approaches are used in preservative challenge testing. These approaches come from the Schülke KoKo Test, United States Pharmacopeia (USP), and the European Pharmacopeia.
Schülke KoKo Test vs. U.S. Pharmacopeia vs. European Pharmacopeia
Pseudomonas aeruginosa, Staphylococcus aureus, Aspergillus niger, and Candida albicans must be tested for in all cosmetic products sold in the European Union. In addition to the microbes above, testing with microbes known to lead to spoilage of cosmetic products is recommended but not required. In the United States, the USP guidelines require testing of Escherichia coli in addition to Pseudomonas aeruginosa, Staphylococcus aureus, Aspergillus niger, and Candida albicans. Spoilage microbes are not needed for PET. In contrast to the pharmacopeia tests, which only evaluate pathogenic microbes, the Schülke KoKo test evaluates product spoiling microorganisms. Schülke KoKo test’s spoiling microorganisms are based on decades of cosmetic testing experience from Schülke & Mayr in Germany.
USP preservative efficacy testing evaluates cosmetic products by exposing them to a single microbial strain at a time. Additional details on USP preservative challenge testing, which follows similar procedures to the European Pharmacopeia, can be found through reading our articles on Preservative Challenge Testing And USP 51, Preservative Efficacy Testing For Medical Devices, and Preservative Challenge Testing For Cosmetic Products. In contrast, for the Schülke KoKo test, the single cultivated microbes are brought together into a mixed suspension to challenge the cosmetic product. A new mixed suspension is prepared for each of the six inoculation cycles for Schülke KoKo testing. Since the Schülke KoKo test uses a mixed microbe inoculation, it simulates microbial exposure of a product during production, filling, and use. Indeed, a product would likely be exposed to multiple microbes at once instead of one at a time. Although different from the USP and European Pharmacopeia methods, the Schülke KoKo Test is a reliable test method for assessing the efficacy of antimicrobial preservation of cosmetic products.
First, cultures of Candida albicans (ATCC No. 10231), Aspergillus brasiliensis (ATCC No. 16404), Escherichia coli (ATCC No. 8739), Pseudomonas aeruginosa (ATCC No. 9027), and Staphylococcus aureus (ATCC No. 6538) are prepared. Stock cultures of these organisms are prepared by centrifuging an ATCC culture removing residual media from the prior culture and resuspending the microorganisms in a sterile suspension fluid. The suspension fluids for each microorganism referenced above are prepared at a microbial count of about 1 × 108 colony-forming units per milliliter (CFU/mL).
Preservative efficacy testing is performed in five sterile, capped containers. Suppose the product’s original container is sterile, can be entered aseptically, and holds an appropriate product volume. In that case, the original filled product containers may be used. Each of the five product samples is injected with a test suspension of either Candida albicans, Aspergillus brasiliensis, Escherichia coli, Pseudomonas aeruginosa, or Staphylococcus aureus. Each product sample is exposed to only a single microbe of the five listed, and all microbes are exposed to the product during testing. The microbial test suspension injected is between 0.5%- 1% of the volume of the product under assessment and is at a concentration of 1 × 105 and 1 × 106 CFU/ml of the product for category 1-3 products. The final concentration per ml of product for category 4 products is between 1 × 103 and 1 × 104 CFU/mL.
Each of the five product samples with their respective microbial injections is incubated at a simulated room temperature of 22.5 ± 2.5°C or 32.5 ± 2.5°C depending upon the microbe the product sample is exposed. Microbial counts are taken at 7, 14, and 28 days for most microorganisms tested. The plate-count method determines the number of CFU present in each of the inoculated product samples. This plate-count method is completed in duplicate. Once all counts are taken, the change in log10 of the concentration of CFU/ml is calculated for each microorganism. The requirements for antimicrobial effectiveness are met if no increase in microbial growth for the various organisms tested occurs within the timeframes specific to each organism. are met. No increase in microbial growth is defined as not more than 0.5 log10 units more than the previous microbial growth value.
How is bacterial endotoxin testing performed?
LAL is dissolved in endotoxin-free water or buffer solution to create a standard endotoxin stock solution without interfering factors. Next, the endotoxin stock solutions are calibrated to the current World Health Organization International Standard for Endotoxin. Endotoxin is expressed in Endotoxin Units (EU). One USP Endotoxin Unit (EU) is equal to one International Unit of endotoxin. Then a serial dilution of the endotoxin standard is prepared. Solutions for samples to be tested are prepared by immersing the device or dissolving the product in the same water or buffer solution used for the endotoxin stock solution. If necessary, the pH of the solution will be adjusted to fall within the range of 6.0-8.0.
A LAL gel-clot technique is used for detecting or quantifying endotoxins based on lysate reagent clotting upon endotoxin exposure. A series of tests are performed to confirm the sensitivity of the lysate solution and for any interfering factors. The sensitivity of the lysate solution is considered to be the minimum concentration of endotoxin required to cause the lysate to clot under test conditions. Both quantitative and limit testing using the LAL gel-clot technique can be performed. For brevity, only the bacterial limit test is described below.
For bacterial endotoxin limit testing of your medical device, four solutions (Solutions A, B, C, and D) are prepared. The details of these solutions are shown in Table 1 of USP 85. Solutions B and C are positive controls with standard endotoxin solution at twice the concentration of the lysate sensitivity. The negative control Solution D is a negative control that consists of water or buffer solution.
A BET test is valid when all Solution B and Solution C samples test positive, and all Solution D samples test negative. In other words, the test is accurate when the positive controls test positive and the negative controls test negative. If both Solution A samples test negative for bacterial endotoxin, the samples pass the BET test limits. If both Solution A samples test positive for endotoxin, the samples fail the BET test. The BET test is repeated if one of the Solution A samples tests positive and the other tests negative. Note that medical device samples do not comply with the BET testing limits if a positive test result is found for one or both Solution A sample replicates.
Amoebocyte lysate reacts to endotoxins and some β-glucans. Amoebocyte Lysates that do not respond to glucans are available. These amoebocyte lysates have the G factor reacting to glucans removed or inhibited. If you are concerned about the presence of glucans in your samples, ensure that an amoebocyte lysate is used. For medical devices, LAL testing has special considerations for product families and special medical device endotoxin limits. There are also endotoxin limits for drugs and biologics. Should your products be above their specified limits, endotoxin controls and depyrogenation studies are available to support reducing a product’s endotoxin concentration. In rare cases, traditional LAL testing may not be possible, and in-vivo pyrogen testing governed by USP 151 may be needed.
What are the differences between preservative challenge and endotoxin testing?
Preservative challenge and endotoxin tests are very different. Preservative challenge testing determines the best preservative to use and the minimum effective concentration needed to preserve multidose parenteral products, topicals products, food, or other healthcare items that contain preservatives. In contrast, bacterial endotoxin testing evaluates product contents for endotoxins. Overall, preservative challenge testing is an evaluation of the antimicrobial properties of a product, while bacterial endotoxin testing is a product safety test.
Summary
Overall, preservative challenge (also known as preservative efficacy testing) and bacterial endotoxin testing are imperative for regulatory approval of parenteral products, topical products, medical devices, and combination products. These tests ensure that medical-grade products are pyrogen-free and have sufficient antimicrobial activity to keep patients safe during product use. Preservative efficacy testing evaluates injectables, topicals, orals, and antacids made with an aqueous base for their antimicrobial activity. In contrast, bacterial endotoxin testing evaluates products for their endotoxin concentrations. All in all, ensure you choose a contract manufacturing organization that can support you with appropriate preservative challenge testing and bacterial endotoxin testing for your unique parenteral product, topical, medical device, or combination product needs.
MycoScience is a contract manufacturing organization specializing in sterile syringe and vial filling cosmetic products or for products used in animal studies. In addition, MycoScience offers testing services, including Preservative Efficacy Testing, Cytotoxicity Testing, Bioburden Testing, Cleaning Validations, Microbial Aerosol Challenge Testing, Accelerated Aging, Microbiology Testing, EO Residual Testing, Bacterial Endotoxin Testing, Package Integrity Testing, Sterilization Validations & Environmental Monitoring services medical devices and allied industries. MycoScience is an ISO 13485 certified facility.
References
Review: Infection, fever, and exogenous and endogenous pyrogens: some concepts have changed. Innate Immunity. August 1, 2004.
Galanos C. and Freudenberg M. A. Bacterial endotoxins: biological properties and mechanisms of action. Mediators of Inflammation. 1993; 2(7): S11–S16.
United States Pharmacopeial Convention. <51> Antimicrobial Effectiveness Testing. Rockville, MD, USA. 2021. (USPC <51>).
United States Pharmacopeial Convention. <85> Bacterial Endotoxins Test. Rockville, MD, USA. 2021. (USPC <85>).
Sharing this in your social netwroks