Top FDA-approved sterile product formulation and filling environments
How does the United States Food & Drug Administration (FDA) define sterile?
Under the strictest definition of sterility, an item or product is sterile when there is the complete absence of viable microorganisms (bacteria, yeasts, viruses, and molds). For regulatory purposes, sterility is defined by acceptance criteria based on calculated contamination probability. An acceptable level of contamination risk for most items is the probability of a single contaminated product out of a million manufactured products. However, sterility criteria may be more stringent or lax depending upon the intended use of the medical device or product. Sterile product testing, regulated by FDA sterile guidances, is used to sterilize and keep product sterile. Activities that require keeping products sterile include syringe filling (using syringe filling machines), vial filling (using vial filling machines), and other sterile product packaging activities.
What does the FDA consider non-sterile processes?
Non-sterile processes are methods or procedures undertaken in an environment where bioburden is controlled to safety levels based on product attributes, route of administration, and target patient population. Non-sterile processes contrast to sterile processes, in which the bioburden is essentially eliminated.
What are examples of common medical products manufactured in non-sterile environments?
- Metered-dose and dry powder inhalants
- Nasal sprays
- Optics
- Topicals
- Oral liquids (aqueous)
- Liquid-filled capsules
- Oral tablets and powder-filled capsules
The non-sterile products listed above are ranked with respect to their potential risk of microbiological contamination (from high to low). The same list applies to medical devices for use in the same body areas (nasopharynx, vagina, skin, rectum, and mouth). Sterile product testing must be performed on non-sterile processes to ensure they meet non-sterile processing microbial criteria.
What are sterile environments, and why are they important?
Sterile product formulation and filling processes must be performed in controlled environments with minimal exposure to microbes, chemicals, or particulates per FDA sterile guidance. The essential sterile product manufacturing activities (including syringe filling and vial filling) are completed in classified environments that conform to the International Organization for Standardization (ISO) procedures. For filling activities, syringe filling machines and vial filling machines are inside ISO cleanrooms. A pressure cascade for classified, side-by-side cleanrooms keeps airflow from cleaner areas to dirtier areas. The lower the number for ISO cleanroom classification, the cleaner the processing environment. Equipment washing occurs in less sterile areas (ISO 7 or ISO 8 classified areas). Non-sterile processing occurs in slightly cleaner ISO 7 or ISO 6 environments, while aseptic processing occurs in ISO 5 environments. ISO 5 environments eliminate the need for human operations (the highest contamination risk) and introduce materials through sterile airlocks and pass-throughs. Table 1 provides some examples of ISO classifications for formulation and filling environments that meet FDA sterile guidance.
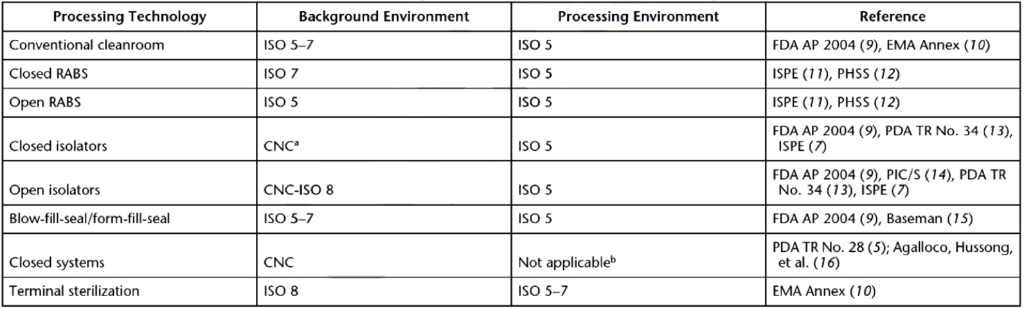
Top FDA-approved sterile product formulation and filling environments
#1: Conventional cleanrooms
In conventional cleanrooms, product processing activities are performed under ISO level 5 conditions and unidirectional airflow. These activities can be supported by ISO 5, ISO 6, or ISO 7 environments with gowned personnel. A limited separation between gowned personnel and sterile materials or product contact surfaces may occur in conventional cleanrooms. Cleanroom decontamination is often performed by personnel for ISO level 5 facilities. Syringe filling machines and vial filling machines may be used in conventional cleanrooms that meet FDA sterile guidances (ISO classifications). However, conventional cleanrooms will not be able to fill sterile products that require the highest level of sterility and sterile product testing.
#2: Restricted access barrier system (RABS)
A standard RABS provides ISO 5 rated unidirectional air within the barrier area. RABS are built within conventional ISO 5–7 cleanrooms. The operation of RABS comes in two flavors. One mode of operation is like an isolator where the operation is closed to personnel. The other allows barriers to open for human manipulation. When humans are involved in operations within the RABS, glove ports are used to handle sterile items, and any material or equipment transfer is achieved without opening the system. If a RABS is opened mid-process, the sterile area becomes the quality level of a conventional cleanroom. Like other ISO-regulated spaces, the RABS must be decontaminated (per FDA sterile guidance) before it is used for product processing, syringe filling, or vial filling. More information on RABS can be found HERE.
#3: Isolators
Isolators are ISO 5 processing environments that keep personnel from the sterile processing environment. A pressure differential is established between the ISO 5 environment and the external environment for sterility. Unidirectional air is not needed in isolators because air overspill functions as an aerodynamic seal when the product exits the isolator into a less clean environment. Isolator decontamination is automated to keep personnel separate from the sterile processing area. Key differences between isolators and RABS can be found HERE.
#4: Blow-fill-seal and form-fill-seal systems
Blow-fill and form-fill sealing systems seal flexible walled containers for sterile products in an ISO 5 environment. Critical filling and sealing activities are performed within a unidirectional airflow environment.
#5: Closed systems
Closed systems encompass isolators and all other processing areas with a complete separation of production materials from human interaction and the surrounding environment. Closed systems vary in complexity from single to multiple-use areas and are either self-sterilized or sterilized before use. Multi-use areas often have multiple modes of ingress and egress and contain numerous vessels or chambers for product processing.
#6: Terminal sterilization systems
Sterile filling systems and terminal sterilization areas for product containers use ISO 7 classified clean areas or better. In these systems, activities are performed within a unidirectional airflow environment. Decontamination of terminal sterilization systems is often performed by personnel.

Summary
Overall, non-sterile and sterile products are processed in ISO-certified clean environments. Non-sterile processes are methods or procedures that are undertaken in an environment where bioburden is controlled to safety levels based on product attributes, route of administration, and target patient population. The criteria for non-sterile products aren’t as stringent as the acceptance criteria for sterile items, where the bioburden is essentially eliminated. Examples of products manufactured in non-sterile environments are dry powder inhalants, nasal sprays, topical products, oral liquids, oral tablets, and powder-filled capsules. Top FDA-approved sterile product formulation and filling environments are 1) conventional cleaning rooms, 2) restricted access barrier systems (RABS), 3) isolators, 4) blow-fill-seal and form-fill-seal systems, 5) closed systems, and 6) terminal sterilization systems. All in all, ensure your contract manufacturing organization has appropriate ISO-certified facilities available for your parenteral product filling needs.
MycoScience is a contract manufacturing organization specializing in sterile syringe and vial filling. MycoScience also offers Preservative Efficacy Testing, Sterilization Validations, Bioburden Testing, Cleaning Validations, Microbial Aerosol Challenge Testing, Accelerated Aging, Microbiology Testing, Cytotoxicity Testing, Bacterial Endotoxin Testing, EO Residual Testing, Package Integrity Testing & Environmental Monitoring services medical devices and allied industries. MycoScience is an ISO 13485 certified facility.
References
Michael J. Akers. Sterile Drug Products Formulation, Packaging, Manufacture, and Quality. Drugs and the Pharmaceutical Sciences. Informa Healthcare. 2010.
United States Pharmacopeial Convention. <1115> Bioburden Control of Non-Sterile Drug Substances and Products. Rockville, MD, USA. 2021. (USPC <1115>).
United States Pharmacopeial Convention. <1211> Sterility Assurance. Rockville, MD, USA. 2021. (USPC <1211>).
Sharing this in your social netwroks